Overview: Understanding electron transport at the molecular level is essential for developing new materials for energy capture and storage applications and next-generation electronic devices. In our group, we use single molecule techniques to directly characterize electron transport in conjugated organic molecules, redox-active molecules, oligomers, and biomolecules such as peptides and protein nanowires.
Intermolecular Charge Transport in Host-Guest Supramolecular Complexes
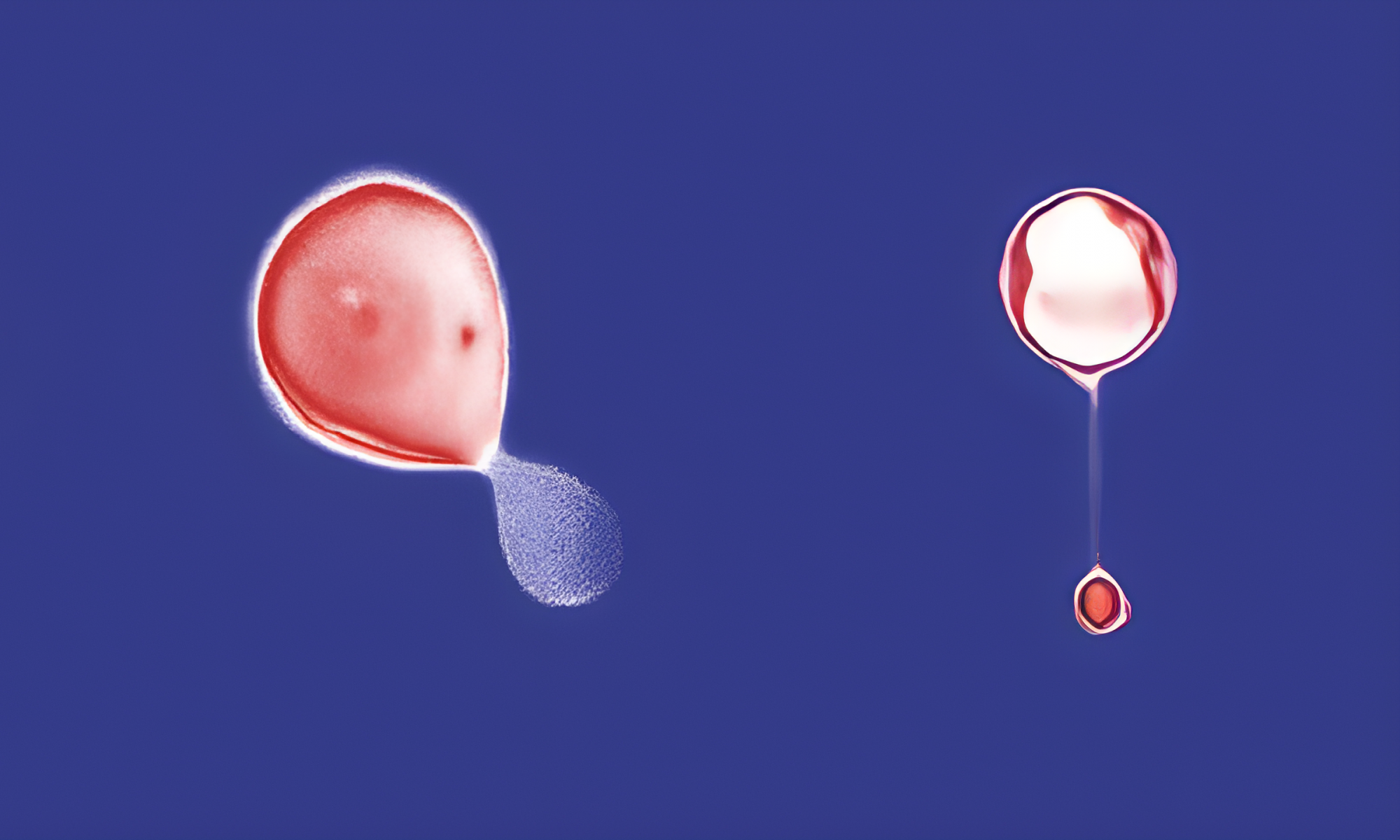
 Intermolecular charge transport through π-conjugated molecules plays an essential role in biochemical redox processes and energy storage applications. In this work, we observe highly efficient intermolecular charge transport upon dimerization of pyridinium molecules in the cavity of a synthetic host (cucurbit[8]uril, CB[8]). Stable, homoternary complexes are formed between pyridinium molecules and CB[8] with high binding affinity, resulting in an offset stacked geometry of two pyridiniums inside the host cavity. The charge transport properties of free and dimerized pyridiniums are characterized using a scanning tunneling microscope-break junction (STM-BJ) technique. Our results show that π-stacked pyridinium dimers exhibit comparable molecular conductance to isolated, single pyridinium molecules, despite a longer transport pathway and a switch from intra- to intermolecular charge transport. Control experiments using a CB[8] homologue (cucurbit[7]uril, CB[7]) show that the synthetic host primarily serves to facilitate dimer formation and plays a minimal role on molecular conductance. Molecular modeling using density functional theory (DFT) reveals that pyridinium molecules are planarized upon dimerization inside the host cavity, which facilitates charge transport. In addition, the π-stacked pyridinium dimers possess large intermolecular LUMO-LUMO couplings, leading to enhanced intermolecular charge transport. Overall, this work demonstrates that supramolecular assembly can be used to control intermolecular charge transport in π-stacked molecules.
Intermolecular charge transport through π-conjugated molecules plays an essential role in biochemical redox processes and energy storage applications. In this work, we observe highly efficient intermolecular charge transport upon dimerization of pyridinium molecules in the cavity of a synthetic host (cucurbit[8]uril, CB[8]). Stable, homoternary complexes are formed between pyridinium molecules and CB[8] with high binding affinity, resulting in an offset stacked geometry of two pyridiniums inside the host cavity. The charge transport properties of free and dimerized pyridiniums are characterized using a scanning tunneling microscope-break junction (STM-BJ) technique. Our results show that π-stacked pyridinium dimers exhibit comparable molecular conductance to isolated, single pyridinium molecules, despite a longer transport pathway and a switch from intra- to intermolecular charge transport. Control experiments using a CB[8] homologue (cucurbit[7]uril, CB[7]) show that the synthetic host primarily serves to facilitate dimer formation and plays a minimal role on molecular conductance. Molecular modeling using density functional theory (DFT) reveals that pyridinium molecules are planarized upon dimerization inside the host cavity, which facilitates charge transport. In addition, the π-stacked pyridinium dimers possess large intermolecular LUMO-LUMO couplings, leading to enhanced intermolecular charge transport. Overall, this work demonstrates that supramolecular assembly can be used to control intermolecular charge transport in π-stacked molecules.
Publication:
H. Yu*, J. Li*, S. Li, Y. Liu, N. E. Jackson, J. S. Moore, C. M. Schroeder, “Efficient Intermolecular Charge Transport in π-Stacked Pyridinium Dimers using Cucurbit[8]uril Supramolecular Complexes”, Journal of the American Chemical Society, 144, 3162-3173 (2022).
Resonant Charge Transport in Molecular Junctions
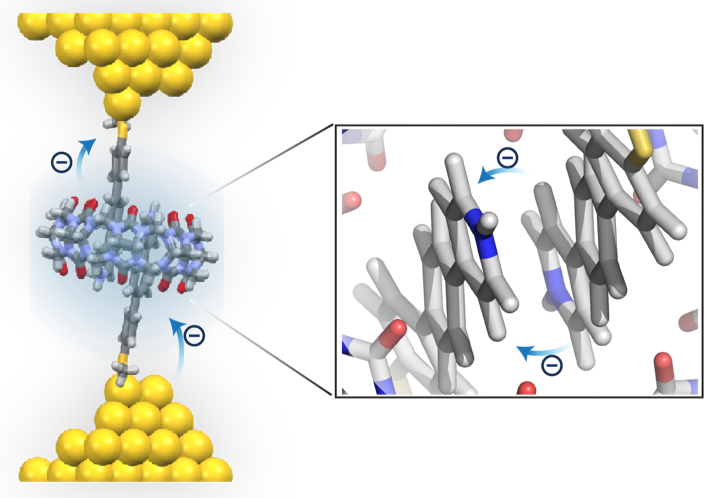
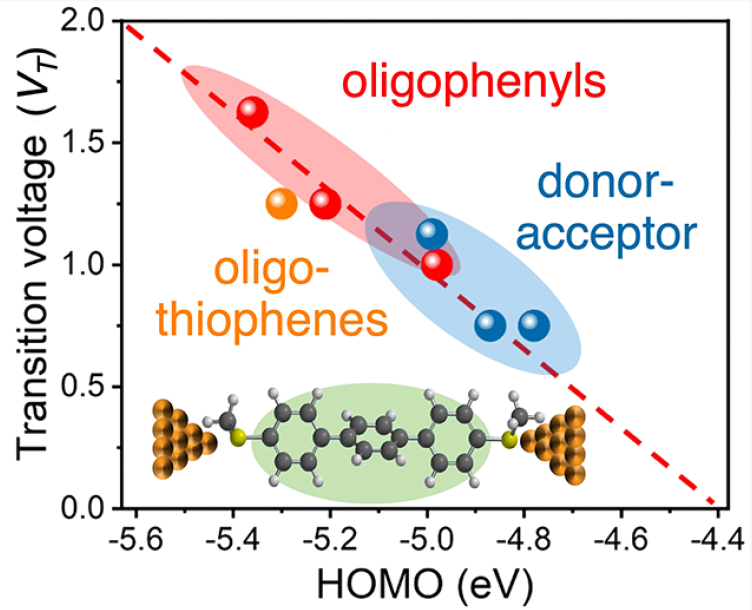 Efficient long-range charge transport is required for high-performance molecular electronic devices. Resonant transport is thought to occur in single molecule junctions when molecular frontier orbital energy levels align with electrode Fermi levels, thereby enabling efficient transport without molecular or environmental relaxation. Despite recent progress, we lack a systematic understanding of the transition between nonresonant and resonant transport for molecular junctions with different chemical compositions. In this work, we show that molecular junctions undergo a reversible transition from nonresonant tunneling to resonant transport as a function of applied bias. Transient bias-switching experiments show that the nonresonant to resonant transition is reversible with the applied bias. We determine a general quantitative relationship that describes the transition voltage as a function of the molecular frontier orbital energies and electrode Fermi levels. Overall, this work highlights the importance of frontier orbital energy alignment in achieving efficient charge transport in molecular devices.
Efficient long-range charge transport is required for high-performance molecular electronic devices. Resonant transport is thought to occur in single molecule junctions when molecular frontier orbital energy levels align with electrode Fermi levels, thereby enabling efficient transport without molecular or environmental relaxation. Despite recent progress, we lack a systematic understanding of the transition between nonresonant and resonant transport for molecular junctions with different chemical compositions. In this work, we show that molecular junctions undergo a reversible transition from nonresonant tunneling to resonant transport as a function of applied bias. Transient bias-switching experiments show that the nonresonant to resonant transition is reversible with the applied bias. We determine a general quantitative relationship that describes the transition voltage as a function of the molecular frontier orbital energies and electrode Fermi levels. Overall, this work highlights the importance of frontier orbital energy alignment in achieving efficient charge transport in molecular devices.
Publication:
S. Li, H. Yu, J. Li, N. Angello, E. R. Jira, B. Li, M. D. Burke, J. S. Moore, C. M. Schroeder, “Transition between Non-resonant and Resonant Charge Transport in Molecular Junctions”, Nano Letters, 21, 8340-8347 (2021).
Reversible Switching of Molecular Conductance using Electrochemical Gating
 Charge transport in electrochemical energy-storage systems critically relies on supporting electrolytes to maintain ionic strength and solution conductivity. Despite recent progress, it is not fully understood how the solvation environment affects molecular charge transport of redox-active species near electrode interfaces. In this work, we characterize the charge-transport properties of bipyridinium molecules in a series of different supporting electrolyte and counterion environments using a combination of experiments and computational modeling. Interestingly, our results show that molecular charge transport in viologens critically depends on the chemical identity of counterions and the solvation environment. Using an electrochemical scanning tunneling microscope-break junction (ECSTM-BJ) instrument, we observe a large and reversible 10-fold enhancement in molecular conductance upon electrochemical reduction of the viologen redox pair (V2+/+) to the radical cationic state in the electrolytic solution. Density functional theory (DFT) simulations show that charge transport is enhanced due to molecular conformational changes and planarization resulting from interactions with different counterions, which ultimately leads to enhanced charge transport in the reduced state. Overall, this work highlights the role of the counterion species on electrochemical charge transport in redox-active molecules that underpin the design of new energy-storage systems or programmable molecular electronic devices.
Charge transport in electrochemical energy-storage systems critically relies on supporting electrolytes to maintain ionic strength and solution conductivity. Despite recent progress, it is not fully understood how the solvation environment affects molecular charge transport of redox-active species near electrode interfaces. In this work, we characterize the charge-transport properties of bipyridinium molecules in a series of different supporting electrolyte and counterion environments using a combination of experiments and computational modeling. Interestingly, our results show that molecular charge transport in viologens critically depends on the chemical identity of counterions and the solvation environment. Using an electrochemical scanning tunneling microscope-break junction (ECSTM-BJ) instrument, we observe a large and reversible 10-fold enhancement in molecular conductance upon electrochemical reduction of the viologen redox pair (V2+/+) to the radical cationic state in the electrolytic solution. Density functional theory (DFT) simulations show that charge transport is enhanced due to molecular conformational changes and planarization resulting from interactions with different counterions, which ultimately leads to enhanced charge transport in the reduced state. Overall, this work highlights the role of the counterion species on electrochemical charge transport in redox-active molecules that underpin the design of new energy-storage systems or programmable molecular electronic devices.
Publication:
J. Li, S. Pudar, H. Yu, S. Li, J. S. Moore, J. Rodríguez-López, N. E. Jackson, C. M. Schroeder, “Reversible Switching of Molecular Conductance in Viologens is Controlled by the Electrochemical Environment”, Journal of Physical Chemistry C, 125, 21862-21872 (2021).
Covalent Ag-C Anchors based on Unprotected Terminal Acetylenes
 Robust molecule-metal linkages are essential for developing high-performance and air-stable devices for molecular and organic electronics. In this work, we developed a facile new method for forming robust and covalent bonding contacts between unprotected terminal acetylenes and metal (Ag) interfaces. Using this approach, we directly characterized the charge transport properties of conjugated oligophenylenes with covalent metal-carbon contacts to silver electrodes formed from unprotected terminal acetylene anchors. We performed single molecule charge transport experiments and molecular simulations on a series of arylacetylenes using gold and silver electrodes. Our results show that molecular junctions on silver electrodes spontaneously form silver-carbynyl carbon (Ag-C) contacts, resulting in a nearly 10-fold increase in conductance compared to the same molecules on gold electrodes. Overall, this work presents a simple, new electrode-anchor pair that reliably forms molecular junctions with stable and robust contacts for molecular electronics.
Robust molecule-metal linkages are essential for developing high-performance and air-stable devices for molecular and organic electronics. In this work, we developed a facile new method for forming robust and covalent bonding contacts between unprotected terminal acetylenes and metal (Ag) interfaces. Using this approach, we directly characterized the charge transport properties of conjugated oligophenylenes with covalent metal-carbon contacts to silver electrodes formed from unprotected terminal acetylene anchors. We performed single molecule charge transport experiments and molecular simulations on a series of arylacetylenes using gold and silver electrodes. Our results show that molecular junctions on silver electrodes spontaneously form silver-carbynyl carbon (Ag-C) contacts, resulting in a nearly 10-fold increase in conductance compared to the same molecules on gold electrodes. Overall, this work presents a simple, new electrode-anchor pair that reliably forms molecular junctions with stable and robust contacts for molecular electronics.
Publication:
S. Li, H. Yu, X. Chen, A. A. Gewirth, J. S. Moore, C. M. Schroeder, “Covalent Ag-C Bonding Contacts from Unprotected Terminal Acetylenes for Molecular Junctions”, Nano Letters, 20, 5490–5495 (2020).
Single Molecule Charge Transport in Sequence-defined Oligomers
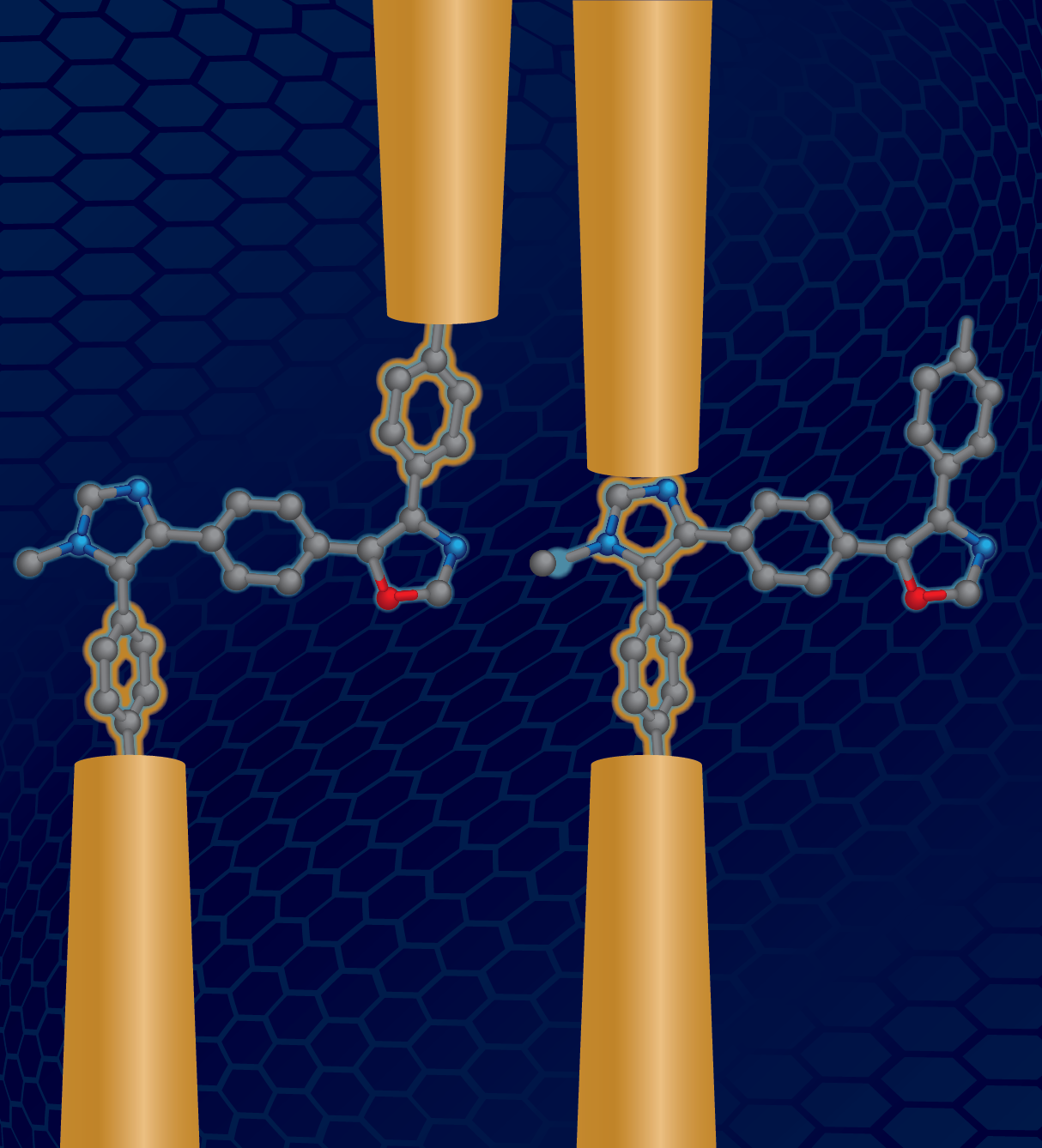 A major challenge in synthetic polymers lies in understanding how primary monomer sequence affects materials properties. In this work, we show that charge transport in single molecule junctions of conjugated oligomers critically depends on the primary sequence of monomers. A series of sequence-defined oligomers ranging from two to seven units was synthesized by an iterative approach based on the van Leusen reaction, providing conjugated oligomers with backbones consisting of para-linked phenylenes connected to oxazole, imidazole, or nitro-substituted pyrrole. The charge transport properties of these materials were characterized using a scanning tunneling microscope-break junction (STM-BJ) technique, thereby enabling direct measurement of molecular conductance for sequence-defined dimers, trimers, pentamers, and a heptamer. Our results show that oligomers with specific monomer sequences exhibit unexpected and distinct charge transport pathways that enhance molecular conductance more than 10-fold. A systematic analysis using monomer substitution patterns established that sequence-defined pentamers containing imidazole or pyrrole groups in specific locations provide molecular attachment points on the backbone to the gold electrodes, thereby giving rise to multiple conductance pathways. These findings reveal the subtle but important role of molecular structure including steric hinderance and directionality of heterocycles in determining charge transport in these molecular junctions. This work brings new understanding for designing molecular electronic components.
A major challenge in synthetic polymers lies in understanding how primary monomer sequence affects materials properties. In this work, we show that charge transport in single molecule junctions of conjugated oligomers critically depends on the primary sequence of monomers. A series of sequence-defined oligomers ranging from two to seven units was synthesized by an iterative approach based on the van Leusen reaction, providing conjugated oligomers with backbones consisting of para-linked phenylenes connected to oxazole, imidazole, or nitro-substituted pyrrole. The charge transport properties of these materials were characterized using a scanning tunneling microscope-break junction (STM-BJ) technique, thereby enabling direct measurement of molecular conductance for sequence-defined dimers, trimers, pentamers, and a heptamer. Our results show that oligomers with specific monomer sequences exhibit unexpected and distinct charge transport pathways that enhance molecular conductance more than 10-fold. A systematic analysis using monomer substitution patterns established that sequence-defined pentamers containing imidazole or pyrrole groups in specific locations provide molecular attachment points on the backbone to the gold electrodes, thereby giving rise to multiple conductance pathways. These findings reveal the subtle but important role of molecular structure including steric hinderance and directionality of heterocycles in determining charge transport in these molecular junctions. This work brings new understanding for designing molecular electronic components.
Publication:
H. Yu*, S. Li*, K. E. Schwieter, Y. Liu, B. Sun, J. S. Moore, C. M. Schroeder, “Charge Transport in Sequence-defined Conjugated Oligomers”, Journal of the American Chemical Society, 142, 4852-4861 (2020).
Quantum Interference in Oxazole-Terminated Oligomers
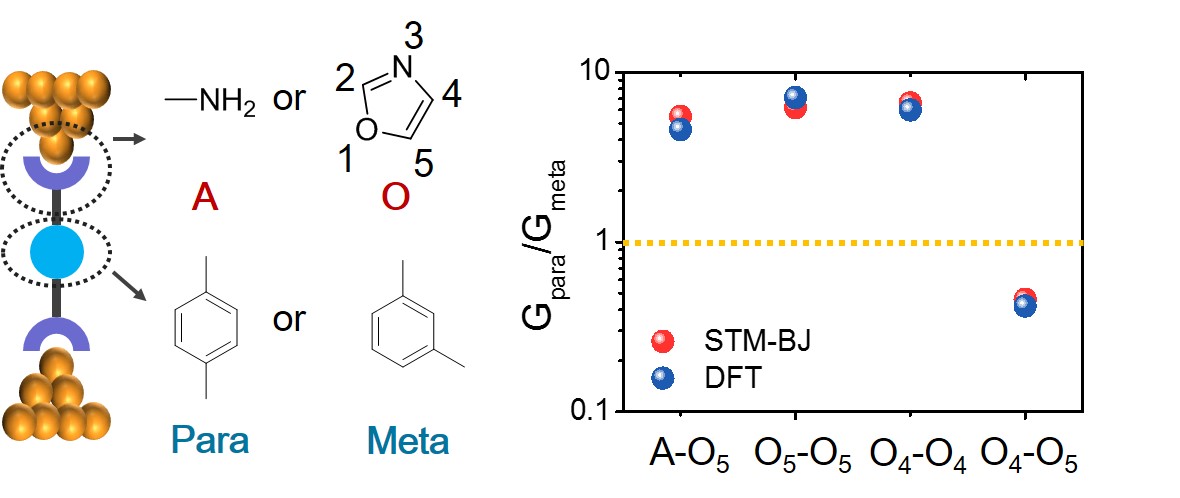 Charge transport in single molecule junctions critically depends on the chemical identity of anchor groups used to connect molecular wires to electrodes. In this work, we report the charge transport properties of conjugated oligomers with oxazole anchors, focusing on the role of the heteroatom substitution position in terminal oxazole groups. Our results show that oxazole serves as an efficient anchor group to form stable gold-molecule-gold junctions. We further observe quantum interference (QI) effects in oxazole-terminated phenylene molecular junctions, including destructive QI in meta-substituted phenyl rings and constructive QI in para-substituted phenyl rings containing terminal oxazole groups with the same chemical constitution on both termini (i.e. O5O5 (5-oxazolyl) or O4O4 (4-oxazolyl) linkages on both termini). Surprisingly, meta-substituted phenyl rings with non-equivalent constitutions (i.e. O4O5 oxazole terminal linkages) show unexpectedly higher conductance compared to para-substituted analogs. These results suggest that charge transport in oxazole-terminated molecules is determined by the heteroatom substitution position of the oxazole anchor in addition to the aryl substitution pattern of the pi-conjugated core. Our results further show that conjugated molecules with homogeneous oxazole linkages obey a quantum circuit rule such that GO4-p-O4/GO4-m-O4 = GO5-p-O5/GO5-m-O5, where G is molecular conductance. Overall, our work provides key insight into the development of new chemistries for molecular circuitry in the rapidly advancing field of single molecule electronics.
Charge transport in single molecule junctions critically depends on the chemical identity of anchor groups used to connect molecular wires to electrodes. In this work, we report the charge transport properties of conjugated oligomers with oxazole anchors, focusing on the role of the heteroatom substitution position in terminal oxazole groups. Our results show that oxazole serves as an efficient anchor group to form stable gold-molecule-gold junctions. We further observe quantum interference (QI) effects in oxazole-terminated phenylene molecular junctions, including destructive QI in meta-substituted phenyl rings and constructive QI in para-substituted phenyl rings containing terminal oxazole groups with the same chemical constitution on both termini (i.e. O5O5 (5-oxazolyl) or O4O4 (4-oxazolyl) linkages on both termini). Surprisingly, meta-substituted phenyl rings with non-equivalent constitutions (i.e. O4O5 oxazole terminal linkages) show unexpectedly higher conductance compared to para-substituted analogs. These results suggest that charge transport in oxazole-terminated molecules is determined by the heteroatom substitution position of the oxazole anchor in addition to the aryl substitution pattern of the pi-conjugated core. Our results further show that conjugated molecules with homogeneous oxazole linkages obey a quantum circuit rule such that GO4-p-O4/GO4-m-O4 = GO5-p-O5/GO5-m-O5, where G is molecular conductance. Overall, our work provides key insight into the development of new chemistries for molecular circuitry in the rapidly advancing field of single molecule electronics.
Publication:
S. Li*, H. Yu*, K. E. Schwieter, K. Chen, B. Li, Y. Liu, J. S. Moore, C. M. Schroeder, “Charge Transport and Quantum Interference in Oxazole-Terminated Conjugated Oligomers”, Journal of the American Chemical Society, 141, 16079 (2019).
Other relevant publications:
- S. Li, J. Li, H. Yu, S. Pudar, B. Li, J. Rodríguez-Lopez, J. S. Moore, C. M. Schroeder, “Characterizing Intermolecular Interactions in Redox-active Pyridinium-based Molecular Junctions”, Journal of Electroanalytical Chemistry, (2020).
- B. Li, H. Yu, E. C. Montoto, Y. Liu, S. Li, K. Schwieter, J. Rodriquez-Lopez, J. S. Moore, C. M. Schroeder, “Intrachain Charge Transport through Conjugated Donor-Acceptor Oligomers”, ACS Applied Electronic Materials, 1, 7 (2019).